How eCTD 4.0 implementation in Regulatory Affairs
eCTD 4.0 – The electronic Common Technical Document (eCTD) is standardized guideline for submitting regulatory
information to health authorities (HAs). It streamlines the process of electronically implementing the Common Technical Document (CTD). An eCTD comprises individual PDF documents organized hierarchically according to the CTD structure. Additionally, it features an XML backbone that interlinks
essential documents and provides submission-related information. The primary objectives behind introducing eCTD were to alleviate the workload for HA reviewers and to establish a universally accepted standard format across regulatory authorities.
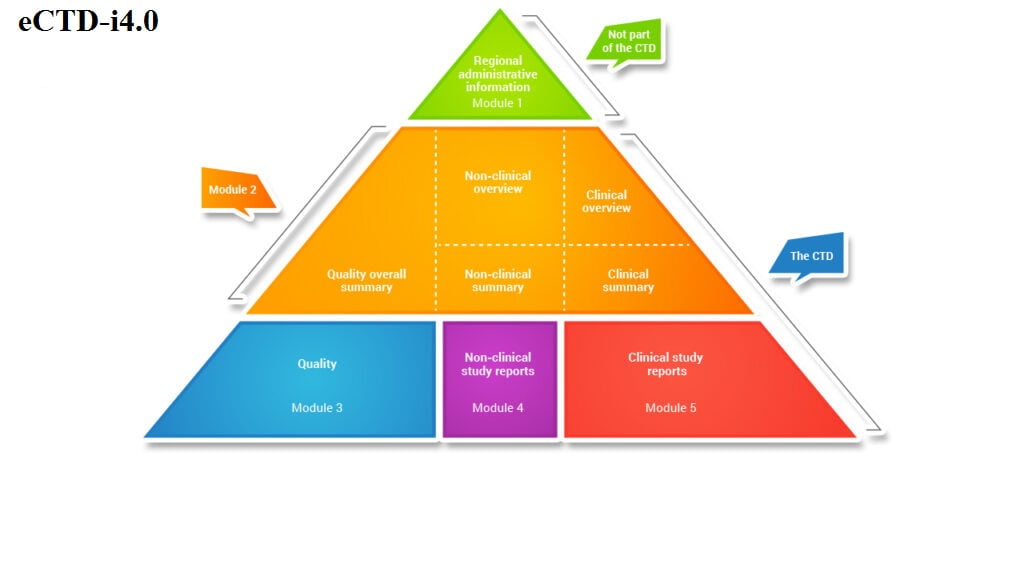
Region-Specific Information: This module contains details specific to the regulatory region where the submission is being made. It includes information relevant to local guidelines, requirements, and specific documentation needed for that region.
Summary Documents: In this module, concise summaries of the entire submission are provided. These
summaries serve as an overview for health authorities (HAs) and reviewers. Key components include the executive summary, product information, and conclusions.
Information Related to Quality: This module focuses on the quality aspects of the submission. It includes data related to the drug substance, drug product, manufacturing processes, stability studies, and analytical methods. Ensuring product quality and safety is paramount in this section.
Non-Clinical Study Reports: Non-clinical study data, such as pharmacology, toxicology, and pharmacokinetics, are documented here. These reports provide evidence of the drug’s safety and efficacy in preclinical studies conducted on animals or in vitro.
Clinical Study Reports (CSRs): The CSRs contain detailed information from clinical trials conducted on human subjects. This module covers study protocols, patient demographics, adverse events, efficacy results, and statistical analyses. CSRs play a crucial role in assessing the drug’s safety and effectiveness.
eCTD submissions are accepted for the following applications implementation
Investigational New Drug (INDs):
These submissions pertain to clinical trials and studies conducted during the development of new drugs. INDs provide essential information about the investigational product, its safety, and proposed clinical trials.
New Drug Applications (NDAs):
NDAs are submitted when seeking approval for a new drug to be marketed in the United States. They include comprehensive data on the drug’s safety, efficacy, manufacturing, labeling, and proposed use.
Abbreviated New Drug Applications
(ANDAs): ANDAs are specific to generic drugs. They demonstrate that the generic version is equivalent to the reference (brand-name) drug in terms of safety, efficacy, and quality.
Biologics License Applications
(BLAs): BLAs are submitted for biological products, including vaccines, blood products, and gene therapies. These applications provide detailed information on the product’s safety, efficacy, and manufacturing processes.
All Applications Following the Submission of the Above-Stated Applications: This category likely includes variations, amendments, and updates related to the initial submissions (e.g., post-approval changes, labeling updates, etc.).
Master Files (MFs): These are part of any of the above-mentioned applications. MFs contain confidential
information related to drug substances, drug products, or manufacturing processes. They are submitted separately and referenced in other applications.
Significant countries, including the US, Europe, Australia, Canada, South Africa, Thailand, and Japan, have
widely adopted the eCTD (electronic Common Technical Document) as the standard format for document submissions due to its manifold advantages over traditional methods. Some of these advantages include:
Automated Sequence Upload: The eCTD allows agencies to automatically upload sequences using its XML backbone, streamlining the submission process.
Enhanced Information Retrieval: Reviewers benefit from hyperlinks within the eCTD, making it easier to navigate and reference relevant information.
Paperless Process: With eCTD, there’s no need to scan, copy, or physically store paper documents, reducing administrative overhead.
Efficient Updates: Changes and updates to dossiers are readily identifiable within the eCTD structure.
Lifecycle Tracking: The eCTD facilitates seamless tracking of a product’s lifecycle, from submission to
approval.
Simultaneous Document Access: Multiple stakeholders can access eCTD documents concurrently, promoting collaboration and efficiency.
In summary, eCTD significantly improves the submission process, benefiting both regulatory agencies and
applicants.