
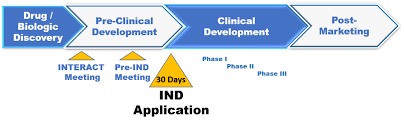
Regulatory Approval Process for Investigational New Drug (NDA-24)
Background Investigational New Drug
Investigational New Drug The drug approval process involves a regulatory pathway through which individuals, organizations, sponsors, or innovators obtain permission to introduce a drug into the market. This process typically includes several stages: applying for clinical trial authorization, conducting clinical trials, applying for marketing authorization, and conducting post-marketing studies. Each country has its own regulatory authority responsible for enforcing rules, issuing guidelines, and overseeing drug marketing regulations.
Investigational Product
An investigational product, as defined by ICH GCP, refers to a pharmaceutical form of an active ingredient or placebo that is being tested or used as a reference in a clinical trial. This can include a marketed product used in a different form than originally approved or used for an unapproved or new indication.
Definition of an Investigational New Drug
According to the Code of Federal Regulations (CFR), an investigational new drug (IND) is defined as a new drug or biological drug used in a clinical investigation. In the United States, an investigational new drug encompasses substances such as drugs, vaccines, or other biological products for which FDA approval is being sought.
Investigational New Drug Application
An Investigational New Drug Application (IND) is a formal request submitted to the Food and Drug Administration (FDA) seeking authorization to administer a new investigational drug or biological product to human subjects. This application is necessary because an investigational new drug, which includes biological products used for diagnostic purposes in vitro, cannot be shipped in interstate commerce without exemption from federal law.
It’s important to note that an IND does not constitute an application for marketing authorization of a drug. Instead, it serves as a notification to the FDA that a sponsor, the entity initiating the clinical investigation, intends to conduct studies involving human subjects after completing preclinical research and data collection.
The IND application, also known as the Investigational New Drug Application (INDA or IND), is a mandatory requirement filed with the FDA to obtain permission for administering a new drug under investigation to human subjects subsequent to the completion of preclinical studies.
The IND process provides safeguards for subjects and ensures that the investigational plan is both effective and designed to meet its specified objectives. The sponsor of the drug, who can be an individual (sponsor-investigator), a pharmaceutical company, a governmental agency, an academic institution, or a private or public organization, assumes responsibility for initiating clinical trials.
The Investigational New Drug Application (INDA or IND) is filed with the FDA under Title 21, Code of Federal Regulations Section 312, which outlines guidelines for the preapproval of all clinical testing.
Content of Investigational New Drug Application
- The requirements for submitting an Investigational New Drug Application (INDA) are outlined in the Code of Federal Regulations and must be accompanied by a cover sheet (Form FDA-1571).
- The necessary information includes:
- Sponsor’s name, address, and contact details.
- Name and title of the individual responsible for monitoring the investigation’s progress.
- Names and titles of those overseeing drug safety evaluation.
- Name and address of any contracted research organization involved.
- Identification of the clinical investigation phase(s).
- Overview of the investigational plan, detailing drug name, active ingredients, formulation, administration route, pharmacological class, study objectives, and duration.
- Description of the investigational plan specifics.
- Reasons for selecting the drug or research study, indications to be studied, evaluation approach, study types, estimated participant numbers, and anticipated risks from animal studies.
- Concise summary of prior drug experience, including reasons for any prior withdrawals.
- Details on chemistry and manufacturing controls, including physical, chemical, and biological characteristics, as well as product stability throughout the clinical investigation.
- Pharmacological and toxicological information.
- If the drug is a combination of previously studied components, include preclinical and clinical data for these components when administered individually and in combination.
- Clinical protocol for the planned study.
- Confirmation that an Institutional Review Board (IRB) has approved the study and will oversee its progress.
- Investigator’s brochure.
- Assurance not to initiate clinical investigations until the IND is effective, signed by the sponsor or authorized representative, along with the date of application signature.
- Following submission to the FDA, the Investigational New Drug (IND) application is reviewed by various divisions within the FDA’s Center for Drug Evaluation and Research.
- The FDA has 30 days from receipt of the IND to determine whether the proposed clinical trial may proceed. If the FDA does not contact the sponsor within this timeframe, the trial may proceed.
- During review, FDA reviewers have the authority to issue a “Clinical Hold” on proposed clinical trials at any time, preventing human testing of the drug. This action may occur for reasons such as:
- Potential threats to the safety of trial subjects, such as illness or injury resulting from treatment.
- Insufficient data to assess risks to patients.
- Investigators not meeting necessary qualifications.
- Misleading or incomplete investigator brochures.
- If all FDA requirements are met, an IND is granted. Once effective, any proposed changes to the original IND must be submitted as amendments for FDA approval thereafter. Regulatory Approval Process
Requirements of IND Application
The FDA places significant regulatory demands on therapeutic drug products, involving extensive laboratory and clinical testing, meticulous sampling procedures, and other resource-intensive processes. Following preclinical testing as per regulations, companies must submit an Investigational New Drug (IND) application to the FDA for approval to proceed with human clinical trials. This application includes a comprehensive plan for studying the drug product and detailed protocols for planned studies. Additionally, the FDA typically requires companies to provide a thorough description of the drug substance, covering its physical, chemical, and biological characteristics, along with details on its preparation method and a list of all components, including inactive ingredients. The IND must detail how the drug substance is composed, manufactured, and controlled, ensuring adequate information on its identification, quality, purity, and strength. Moreover, the IND must include comprehensive pharmacological and toxicological data from animal studies and other tests, demonstrating the sponsor’s conclusion that it is reasonably safe to proceed with clinical trials. During clinical trials, sponsors must adhere to strict reporting requirements, including timely progress reports and immediate notification to the FDA and clinical investigators of any unexpected serious side effects or injuries.