
Analytical Methods Validation for Quality Assurance and Process Validation Experts
Abstract:Analytical Methods Validation
Method validation is a fundamental process in the pharmaceutical industry, essential for ensuring that analytical procedures used in testing are appropriate for their intended purposes. Validation data serve to confirm the performance, consistency, and reliability of these methods. This paper outlines the necessary requirements for method validation and data generation, providing evidence of suitability for specific applications. It also reviews key validation parameters according to ICH guidelines and various pharmacopeias, emphasizing that a method must be fully validated before it is used for release or stability testing. Additionally, it highlights the importance of a change control program to assess the impact of any modifications to validated parameters. The paper distinguishes between method validation, verification, and transfer. Its goal is to offer guidance to Quality Assurance (QA) and Validation Professionals for evaluating method validation data provided by analytical scientists, rather than conducting the validation themselves.
Introduction:Analytical Methods Validation
In the pharmaceutical and biotechnology sectors, companies are increasingly developing and commercializing complex formulations to address medical needs. Maintaining the quality of active pharmaceutical ingredients (APIs) and drug products remains a challenge, requiring effective analytical methods for both release and stability testing. Consequently, method validation is crucial. Quality assurance and validation professionals must assess the validation status of analytical procedures and the impact of any changes to these methods, especially when aiming to streamline operations and expedite time to market.
Numerous publications have detailed method validation throughout the drug development process. This paper aims to guide QA and Validation Engineers specifically on method validation, focusing on chromatographic procedures for chemical assays and impurity testing of small molecule drug products.
Content Overview:

- Review of Guidelines: This section examines various guidelines relevant to the validation of analytical procedures for monitoring pharmaceutical products.
- Analytical Testing and Quality Systems: It explores how analytical testing supports pharmaceutical quality systems.
- Key Validation Parameters: It outlines essential validation parameters that define the characteristics of analytical procedures.
- Analytical Lifecycle Management: A discussion on managing the analytical lifecycle of methods.
- System Suitability Testing: An overview of method performance through system suitability testing.
Regulatory Expectations for Analytical Methods Validation
Method validation is a crucial component of Good Manufacturing Practices (GMPs). The Food and Drug Administration (FDA) and global organizations like ICH and WHO provide various guidelines on this topic. While regulations from the US, Europe, and Japan generally align with ICH guidelines, specific details are not reiterated here.
According to Section 21 of the Code of Federal Regulations (CFR) 211.165(e), “The accuracy, sensitivity, specificity, and reproducibility of test methods employed by the firm shall be established and documented.” This validation and documentation are governed by Section 211.194(a), which relates to Laboratory Records. This regulation is intentionally broad to allow manufacturers to create more specific standard operating procedures (SOPs) tailored to their needs.
In November 2005, the ICH released the updated Q2(R1) guideline, merging ICH Q2A and Q2B to provide comprehensive instructions on validation parameters and procedures. This guideline complements the 21 CFR 211 regulations and enhances consistency in method validation within the pharmaceutical industry. It focuses primarily on separation techniques, which are crucial for quantifying individual components in complex mixtures and includes guidelines on both quantitative and qualitative assays. Furthermore, the ICH guideline emphasizes method robustness, which is the method’s ability to maintain performance despite deliberate variations in operational parameters. In June 2018, the ICH acknowledged the need for updates and proposed developing Q2(R2) to cover additional analytical technologies like spectroscopy and spectrometry, and Q14 to address analytical procedure development, indicating potential future changes.
In July 2015, the FDA revised its guidance document on “Analytical Procedures and Methods Validation of Drugs and Biologics.” This updated document condenses the previous ICH Q2-based guidance, removing much of the ICH content and replacing it with references to ICH guidelines. It recommends considering the intended purpose and scope of the analytical method when selecting instrumentation and methodology. The revised guidance also includes a section on analytical method development, explaining how method parameters are chosen based on their intended use. Additionally, it references the US Pharmacopeia (USP) General Chapters on Validation of Analytical Procedures (<1225>), Verification of Compendial Monographs (<1226>), and Transfer of Analytical Methods (<1224>).
The validation parameters outlined in ICH Q2(R1) and USP <1225> are similar, although robustness testing is not part of USP <1225> validation but is recommended during method development. Robustness involves studying the effects of small, deliberate changes to optimized method conditions, often using experimental designs. This testing is sometimes considered part of pre-validation rather than formal validation.
While the method categories in both guidelines are largely similar, USP <1225> also includes procedures for measuring product performance, such as dissolution testing for solid dosage forms. Most of these guidelines are tailored to traditional dosage forms of small molecule APIs.
Validation Parameters and Their Impact on the Analytical Procedure
Method Development
When developing an analytical procedure, it is crucial to collect comprehensive information before initiating the method development phase. This information should encompass known chemical structures, analyte concentration, solubility, stability, sample matrix, and spectroscopic properties, along with other physical and chemical characteristics of the active pharmaceutical ingredient (API) and other relevant components. Understanding the intended purpose of the method and its suitability for the intended use is fundamental.
Key considerations during the method design phase include the complexity of sample preparation, selection of analytical instrumentation, analysis duration, availability of reagents and standards, and method efficiency in a routine Quality Control (QC) environment. Additionally, factors such as the cost of solvents, materials, and waste management should be taken into account. Evaluating sample throughput is also essential for optimizing method performance.
A detailed white paper outlines a straightforward five-step process for developing a High-Performance Liquid Chromatography (HPLC) method for assay and impurity analysis.8 The data generated during method development will form the basis for establishing validation parameters and defining appropriate acceptance criteria.
Specificity in Method Validation
Specificity is a crucial parameter in method validation, but it is not always explicitly determined during the validation process itself. It is essential to ensure that the method can accurately identify the analyte even in the presence of other substances that might be present. Specificity is vital for detecting issues like unidentified HPLC peaks found during stability testing of biobatch or validation lots, which should be identified and evaluated.
Key Considerations for Specificity:
- Filtration Validation: Filters used for filtering dissolution aliquots must be validated to ensure they do not cause loss of the analyte or interact with the drug product in a way that affects the results.
- Factors Affecting Specificity: Several factors can impact the specificity of a method and may necessitate additional studies or revalidation. These factors include the intrinsic stability of the active pharmaceutical ingredient (API), interactions with excipients, changes in the manufacturing process, composition of the dosage form, raw materials, packaging components, and storage or handling conditions. Any of these factors can lead to unintended changes in the final product and the formation of degradation products.
Specificity Studies for Stability Testing:
For methods monitoring product quality over time, specificity studies often involve forced degradation and stress testing. For example, products stored at room temperature should be subjected to stress testing with drug substance and drug product samples exposed to high temperatures (>40°C), high humidity (>75%), and light. Stress testing also includes exposing the drug substances to acidic, basic, and oxidative conditions. The objective is to achieve an approximately 5-20% loss of active ingredient under the most stringent conditions. For products stored under refrigerated or frozen conditions, stress studies should be evaluated on a case-by-case basis.
Stability-Indicating Methods:
The analytical method must be stability-indicating when testing finished product stability samples. This means the method should accurately measure all main components, impurities, and potential degradation products without interference from the placebo or the matrix. For methods relying on separation techniques, such as chromatography or electrophoresis, peak purity must be demonstrated using appropriate orthogonal detection methods, such as photodiode array or mass spectrometry.
Precision
Precision refers to the degree of consistency in repeated measurements under the same conditions across the operating range of concentrations. For High-Performance Liquid Chromatography (HPLC) used for assay and impurity analysis, precision is assessed at multiple levels, as illustrated in Figure 1.
Levels of Precision:
- Repeatability: This is the variability observed when measuring the same sample multiple times under identical conditions. It includes:
- System Precision: This involves injecting the same sample preparation (usually at 100% concentration) multiple times to assess the reliability of the analytical system.
- Method Precision: This involves preparing and analyzing the same sample multiple times (typically six times) to evaluate both the precision of the analytical system and the variability introduced during sample preparation.
- Intermediate Precision: This measures variability when samples are analyzed by different analysts, on different days, and using different sets of instruments. If a validation was conducted several years ago, it may not have included this parameter, and thus the method might not meet current precision requirements.
- Reproducibility: This assesses the variability when samples are analyzed across different laboratories. Method transfer studies, where the same set of samples is analyzed by both the transferring and receiving laboratories, can demonstrate method reproducibility. Various approaches for conducting method transfers are outlined in the USP General Chapter – Transfer of Analytical Methods <1224>.
By evaluating these levels of precision, the robustness and reliability of the HPLC method can be thoroughly assessed to ensure consistent and accurate results.
Accuracy
Accuracy refers to how close a method’s measurement is to the true value within the working range of concentrations. To determine accuracy, several approaches can be employed:
- Testing Against a Reference Standard: This involves using a reference standard to measure accuracy. The method’s results are compared against the known value of the reference standard to assess how close the measured value is to the true value.
- Complex Samples: Accuracy can also be tested by spiking the active ingredient into a placebo, which is a mixture of excipients. Comparing results with and without the placebo helps to determine if the placebo interferes with the measurement of the component of interest.
- Standard Addition: This technique is used when the sample already contains a small amount of the component of interest. For example, in methods measuring residual solvents, standard addition involves adding known quantities of the analyte to the sample and measuring the change to determine accuracy. This method is also useful when a clean matrix or placebo is not available.
- Comparison with a Reference Method: When a validated reference method exists, accuracy can be assessed by comparing test data obtained from the new method with data from the reference method. This comparison helps validate that the new method provides results consistent with the true value.
Typically, external standardization is used for accuracy calculations, and both linearity and precision must be demonstrated alongside accuracy over the working concentration range.
Detection Limit/Quantitation Limit (DL/QL)
The Detection Limit (DL) and Quantitation Limit (QL) are critical parameters when quantifying low levels of a component, such as impurities or degradation products. These limits can be determined by:
- Signal-to-Noise Ratio: Calculating DL and QL based on the ratio of the signal to noise can provide an estimate of the lowest concentration that can be reliably detected and quantified.
- Standard Deviation and Slope: Alternatively, DL and QL can be calculated using the standard deviation of the response and the slope of the calibration curve.
For methods assessing impurities or degradation products, it is essential to verify the QL within the system suitability testing section to ensure method performance meets the required standards.
Ruggedness and Robustness
Ruggedness
Ruggedness, also known as intermediate precision, evaluates how a method performs under normal variations that might occur during its use. This includes variability introduced by different analysts, instruments, or environmental conditions. Typically, ruggedness is assessed as part of the precision study, specifically focusing on intermediate precision.
Robustness
Robustness measures how a method performs when subjected to small, deliberate changes in its parameters. Unlike precision, which looks at typical variations, robustness examines the method’s stability under controlled alterations. Common factors tested for robustness include:
- Flow rate variations (e.g., ±20%)
- Column length adjustments
- Changes in mobile phase composition
- pH variations
- Detector wavelength shifts
- Sonication time adjustments
These parameters can be studied individually or through experimental designs. Robustness testing also involves evaluating all system suitability parameters. Changes in sample preparation methods, such as shaking time, extraction time, and filtering, should also be assessed.
Solution stability is a crucial aspect of robustness testing. It involves evaluating the stability of all solutions used in the analytical procedure, including standards, samples, system suitability solutions, mobile phases, and reagents. Companies should implement a standard operating procedure (SOP) for the storage of reagents and solvents, including establishing appropriate ‘use-by’ dates. Robustness testing should ensure that the method consistently elutes and quantifies impurities and potential degradation products.
Content of the Analytical Method
The FDA’s 2018 guidance document outlines the essential components that should be included in an analytical method. These sections are:
- Principle/Scope: The fundamental principle behind the method and its intended scope of application.
- Apparatus/Equipment: Details of the equipment and apparatus used in the method.
- Operating Parameters: Specifications for the operational settings and conditions.
- Reagents/Standards: Information on reagents and standards used in the procedure.
- Sample Preparation: Procedures for preparing samples for analysis.
- Standards Solution Preparation: Instructions for preparing standard solutions.
- Procedure: The detailed steps of the analytical procedure.
- System Suitability: Criteria and tests to ensure the system is functioning correctly.
- Calculations: Methods for calculating results from the analytical data.
- Data Reporting: Guidelines for reporting the data obtained from the analysis.
- Reference Standards and Materials: Details on reference standards and materials used for validation and calibration.
Reference Standards and Materials
Reference standards are essential for accurate identification and quantitation in High-Performance Liquid Chromatography (HPLC) analyses. Each organization should have a standard operating procedure (SOP) that governs the qualification, distribution, and maintenance of these reference standards.
Reference standards must be thoroughly characterized, and their impurity profiles must be identified and monitored through appropriate testing. Companies can either use commercially available reference standards or qualify their own secondary standards for use in volumetric testing.
Performance of the Chromatographic Method
System suitability for chromatographic methods is typically established in accordance with USP General Chapter <621>. Validation data are utilized to set the acceptance criteria for system suitability testing (SST). It is advisable to design SST protocols using commercially available reagents, solvents, or standards to ensure consistency and reliability.
When transferring methods to a Quality Control (QC) or contract laboratory, it is crucial to thoroughly evaluate the critical parameters of the methods to ensure they perform as expected in the new setting. Regular evaluation and trending of SST data are recommended to identify potential deviations and prevent system failures.
Validation by Phases
Global guidelines generally require that full method validation be completed before testing the first registration batch of a drug. While comprehensive validation is necessary in later phases of drug development, the expectations for method validation are more flexible during early development stages. This phased approach reflects the evolving nature of analytical methods and the associated risks.
Early Development Phases:
- Flexibility in Validation: During the early stages of drug development, analytical methods are often still evolving due to frequent changes in the drug substance manufacturing process and drug product formulation. As a result, methods may be updated and re-validated regularly to ensure their suitability for intended use. Validation requirements are typically less stringent in these phases due to the smaller number of samples and the fact that testing is conducted by a limited number of analysts in a single laboratory.
- Risk-Based Approach: Validation activities in early development are guided by a risk-based approach, focusing on critical parameters that are essential for the release and monitoring of clinical supplies. This approach ensures that the analytical methods remain fit for purpose despite the ongoing changes in the drug product and manufacturing process.
Late Development Phases:
- Complete Validation Required: As the drug product formulation and manufacturing process stabilize in later development phases, comprehensive method validation becomes necessary. By this stage, the method must be fully validated to meet stringent regulatory requirements, ensuring robustness and reliability for larger-scale production and registration purposes.
Typical Flow of Validation Parameters:
Figure 2 illustrates the typical flow of validation parameters that should be studied based on the phase of product development. In summary, the validation process evolves from a more flexible approach in the early phases to a rigorous validation requirement as development progresses, ensuring the method’s reliability and accuracy for final product release and monitoring.
Changes to the Analytical Methods
Regulatory guidance emphasizes that validation is an ongoing process, making the validation report a dynamic, living document. It is crucial that any analytical procedure used for specific tests is demonstrated to be suitable for its intended purpose through thorough validation. Methods must be fully validated before they are routinely used in Quality Control (QC) laboratories for release or stability testing.
When changes occur in the method or test sample that fall outside the original scope of the validated method, a review of the validation is necessary to ensure the method remains effective. These changes, whether they involve the analytical procedure or test samples, should trigger a re-evaluation of the method’s performance. Changes are managed through a change control program, which tracks and characterizes the modifications.
Examples of Changes Requiring Revalidation:
Table 2 outlines specific changes that may necessitate method revalidation. These might include:
- Changes to the analytical instrumentation or equipment.
- Modifications in the sample preparation procedures.
- Alterations in the reagents or standards used.
- Variations in the analytical conditions, such as flow rates or column types.
- Updates in regulatory or compendial requirements.
Change Control Program:
The change control program evaluates each modification for its potential impact on method validity. It includes:
- Assessing deviations and tracking corrective and preventive actions.
- Reviewing and adjusting the method performance as needed.
- Integrating risk assessment into the revalidation process to ensure the method continues to meet its intended use.
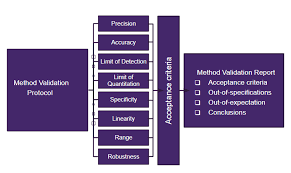
Validation Protocols:
All validation activities—whether initial validation, revalidation, or verification—should be conducted under an approved protocol with defined acceptance criteria. This ensures the analytical method remains fit-for-purpose and compliant with regulatory standards.
Evaluation of Changes:
If an evaluation determines that a change is within the original scope of the method, additional validation may not be required. Such evaluations and their conclusions should be thoroughly documented. Additionally, system suitability test parameters should be reviewed and updated if necessary to maintain the method’s performance and compliance.
Verification of Compendial Monographs
For standard methods recognized by authoritative bodies such as The American Society for Testing and Materials (ASTM), International Organization for Standardization (ISO), and the United States Pharmacopeia (USP), which have already been validated by the method’s sponsor, a full validation is not required by subsequent users. Instead, verification is necessary to ensure the method’s suitability for the test sample.
Verification Requirements:
- USP General Chapter <1226> provides guidance for verifying compendial methods. Verification involves evaluating the analytical method to ensure it is appropriate for the specific material being tested. The verification process should address system suitability requirements under actual conditions of use.
- Key Parameter – Specificity: The most crucial aspect of verification is specificity. It ensures that the method can accurately identify and quantify the material of interest, accounting for all potential impurities and degradation products. This step is vital to confirm that the method is appropriate for the specific material being analyzed.
Adjustments and Documentation:
- Method Adjustments: In practice, the compendial method might need to be adjusted for use in routine analytical laboratories. These adjustments ensure that the method is practical and effective under actual laboratory conditions.
- Verification Plan and Report: The verification process should be conducted under a structured protocol, and the results should be documented in a verification report. This report will detail the verification data and confirm that the method is suitable for the intended use with the specific material.
In summary, while compendial methods do not require revalidation by subsequent users, they must undergo verification to confirm their applicability to specific materials and testing conditions.
Method Lifecycle Management
Analytical methods play a crucial role in determining the quality of drug substances and products. Consequently, the performance of these methods must be continuously monitored and reviewed. Method validation is not a one-time event but an ongoing process that ensures the method remains effective throughout its lifecycle.
Lifecycle Overview:
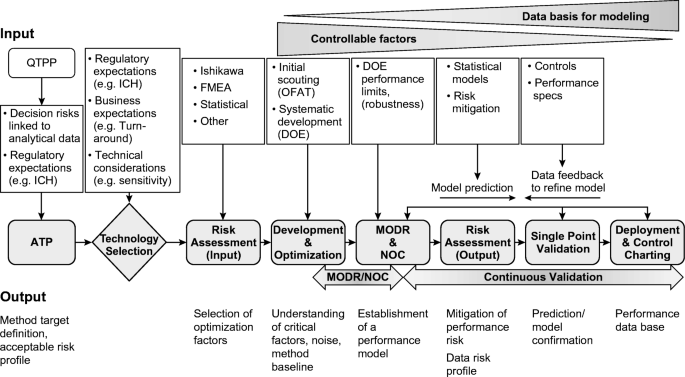
- Development and Validation: The lifecycle begins with the development of the analytical procedure, followed by qualification and validation based on the method’s intended use. Figure 3 illustrates this initial phase, emphasizing that once implemented, the method must undergo periodic reviews and optimization to maintain its robustness over time.
- Ongoing Performance Monitoring: To support effective product testing, it is essential to regularly study the validation of the method to understand its critical parameters. Method performance should be tracked, and trend analyses should be conducted at regular intervals. This proactive approach helps to identify and mitigate any unintended changes that might affect the monitoring of the drug product. Table 3 provides a template for planning validation activities for analytical procedures.
ICH Q12 Framework:
- Regulatory and Technical Considerations: ICH Q12 offers a comprehensive framework for managing the lifecycle of pharmaceutical products using a risk-based and science-based approach. This guideline supports flexible regulatory strategies for post-approval changes, utilizing information gathered during product development.
- Lifecycle Stages: The product lifecycle encompasses three stages:
- Process Development: Establishing the product profile and identifying critical quality attributes.
- Risk Management: Developing the design space and control strategies for the product.
- Lifecycle Management: Ensuring continual improvement and method performance through ongoing verification and adjustment.
Application to Analytical Methods:
Figure 4 illustrates the application of the lifecycle concept to analytical method validation. This framework ensures the maintenance of method performance through development, qualification, and continual verification. During development, the method’s critical attributes are identified with a focus on an analytical target profile. Once the method parameters are established, a validation protocol is used for qualification, and the results are documented in a validation report. After successful validation or verification, the method must be continuously managed to ensure it remains fit for its intended purpose.
Adjustment and Development:
As new information or technologies emerge, or if there are changes to the product target, additional method development or validation may be required. Periodic reviews of method performance are essential to anticipate and address potential issues. Maintaining a sufficient quantity of aged samples is also critical for comparative studies and supporting the method validation lifecycle.
In summary, method lifecycle management involves ongoing validation and performance monitoring to ensure that analytical methods continue to meet their intended purpose throughout the product’s lifecycle.
Audit and Inspection
Analytical methods are essential tools for monitoring the quality of drug substances and products, making their validation crucial to ensure reliability. Method validation is a key focus during quality audits and inspections, where the validation report is thoroughly examined to confirm compliance with industry standards. Table 4 provides an example of how to assess a method validation report for a solid formulation, including validation parameters and recommendations from ICH Q2(R1). This example can be adapted to fit different types of methods or specific company Standard Operating Procedures (SOPs).
Manufacturers should regularly review their validation activities, especially if the activities were conducted before 2012. It is also important to review change control processes, as accumulated minor changes over time might subtly affect the performance of the analytical procedure.
Summary
Analytical procedures are crucial for assessing product quality, including evaluating the potency of finished products and detecting unknown impurities. Therefore, the validation of these procedures is a vital part of pharmaceutical drug manufacturing. Manufacturers must maintain a rigorous validation process to prevent issues that could lead to product investigations or recalls. Effective management of the method validation lifecycle is essential to track and trend any changes to the analytical method, as unintended changes may pose risks or unknown dangers to patient safety.