
EU IDMP Implementation Guide -2_Data elements for the electronic submission of information on medicinal products for human use
EU IDMP Implementation Guide -2_Data elements for the electronic submission of information on medicinal products for human use, The EMA released version 2.0 (EU IG v2.0) in February 2021. This version is primarily designed to assist the European medicines regulatory network in preparing for Step 1. In addition to the content from version 1, it includes:
- Key principles of the target operating model for submitting and maintaining medicinal product data in the EU.
- Additional guidance on populating PMS data elements.
- The foundation for medicinal product data exchange in the EU.
The EMA intends to publish a third version (EU IG v3.0) to aid in the implementation of Step 2 and to incorporate the most recent agreements and details.
The EU IG v2.0 primarily facilitates the execution of Step 1 and is essential for preparing data submission through the PMS for medicinal products approved via the centralised procedure. It offers:
- Clarification on the target operating model concepts of PMS.
- Clarification on the processes for submitting, exchanging, or validating medicinal product information.
- Detailed guidance on registration requirements and information on product data access.
- Clarification on the data elements for electronic submission of human medicinal product information and the relevant business rules.
- Practical examples to help SPOR users accurately structure their product data in scenarios where directly
The information in EU IG v2.0 equips the European medicines regulatory network to get ready for submitting data on all human medicinal products authorized in the EU. It lays the groundwork for practical preparation activities, including:
- Conducting proofs-of-concept for end-to-end processes involving the generation and submission of FHIR messages, validation, and interaction with eCTD.
- Testing various use cases.
The European medicines regulatory network has established a phased release plan for the EU Implementation Guide. The EMA will issue minor versions (EU IG v2.1 and EU IG v2.2) in 2021 to incorporate the latest agreements and details, thereby improving quality.
Substance and product data management services: EU IDMP Implementation Guide
The PMS and SMS will oversee two of the four domains in the SPOR master data categories essential for pharmaceutical regulatory processes: substances, products, organizations, and referentials. SPOR data management services ensure the dependable and uniform exchange of medicinal product information.
These services are crucial for the adoption of ISO IDMP standards within the EU and EEA. The PMS and SMS are built upon the data foundations established by the Referentials Management Service (RMS) and Organizations Management Service (OMS), both of which were introduced by the EMA in June 2017.
The implementation of PMS and SMS is carried out in iterative steps. The initial phase addresses a subset of ISO IDMP data fields, with subsequent phases aiming to fully implement the standards in the EU.
In 2019, the first phase of the SMS allowed users to request new substance term registrations or updates to existing terms via the EMA Service Desk, facilitating EMA’s management of substance data. Future phases will integrate SMS with the European Substance Reference System (EU-SRS) database and develop a user interface for SMS.
The initial phase of PMS will focus on a subset of the ISO IDMP standards for authorized medicinal products. This phase will introduce the new ISO IDMP compatible data submission format, HL7 FHIR, replacing the current eXtended EudraVigilance Product Report Message (XEVPRM).
Future PMS iterations will implement other product data elements of the authorised medicinal product and the investigational medicinal product part of the ISO IDMP standards.
Data standard for information exchange: EU IDMP Implementation Guide
The draft international messaging standard, Fast Healthcare Interoperability Resources (FHIR, pronounced “fire”), has been approved as the foundation for the application programming interface (API) for the Product Management Service (PMS). FHIR will serve as the data standard facilitating the exchange of information regarding medicinal products, substances, and related reference data within the European medicines regulatory network.
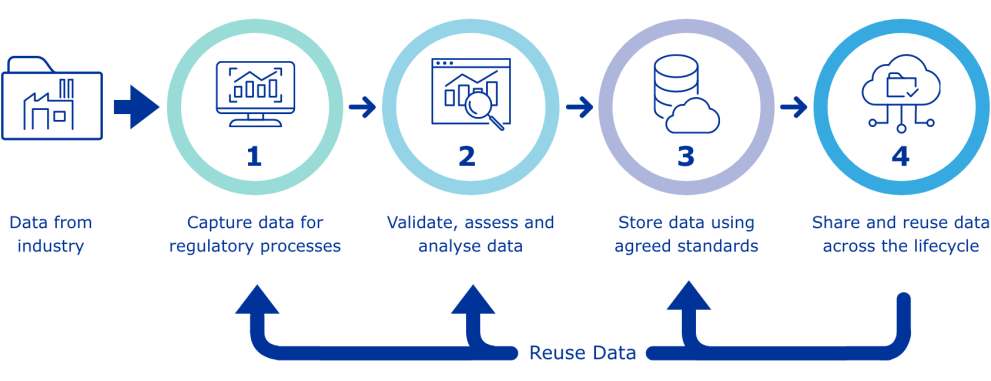
The data-centric target operating model (TOM) illustrates how product data can be reused in regulatory processes and applications. The European Medicines Agency (EMA) is working to align the technical components and business processes with regulatory activities to ensure data quality and consistency across the PMS, national data systems, and pharmaceutical companies’ data systems.
EMA’s electronic application forms (eAF) streamline the capture of data for regulatory processes. The agency is updating these forms to enhance the process of entering product data. Authorized product data that already exists can be retrieved from the PMS database.
The pertinent regulatory body evaluates and oversees the information provided in the dossier (including the application form). Utilizing standardized product data streamlines this evaluation process. Product data and pertinent documents are archived and accessible through the Product Management System (PMS). Upon complete implementation, data and documentation within the PMS will meet the legal mandates outlined in Article 57 of Regulation 726/2004. Authorized product data and documents housed in the PMS can be utilized again for subsequent regulatory interactions between regulatory authorities and pharmaceutical firms.
PMS contribution to TOM in 2022
In 2022, EMA has initiated a shift towards replacing the current PDF-based electronic application forms with web-based counterparts, as part of their TOM implementation efforts. The first form undergoing this transformation is the variation eAF, with plans to extend this update to other forms supporting various regulatory procedures, such as initial marketing authorizations, in subsequent phases.
The PMS is playing a crucial role in facilitating the adoption of electronic forms by providing ISO IDMP-compatible product data for both centrally authorized products (CAPs) and non-centrally authorized products (non-CAPs). Additionally, efforts are underway to enhance the technical infrastructure necessary for accurately reflecting authorized data within the PMS.
Given the intricacies involved in developing and integrating a data-centric business process aligned with regulatory activities and TOM technical components, EMA is adopting an iterative approach to implementation. Consequently, there will be transitional periods during which multiple systems will operate in parallel, despite the inconvenience to regulators and applicants.
EMA is currently engaged in updating its three-year portfolio objectives to inform the portfolio roadmap. This includes prioritizing the implementation roadmap for ISO IDMP, which involves replacing the Article 57 data submission format. These initiatives are being carried out using an agile methodology, with close collaboration between EMA, industry stakeholders, and partners in the European medicines regulatory network to ensure strategic alignment and address the needs of all stakeholders.
In 2022, the EMA aims to advance several initiatives including:
- Ensuring ISO IDMP-compatible product data is accessible for all authorized medicinal products in the EU, covering both centrally and non-centrally authorized products. This involves migrating data and continually updating from EMA’s SIAMED and xEVMPD (Article 57) databases to the PMS in accordance with ISO IDMP standards.
- Facilitating pharmaceutical companies to rectify and enhance PMS product data.
- Allowing data endorsed within a regulatory application to be stored in the PMS.
- Maintaining sufficient data quality in the PMS to ensure confident reuse across procedures.
Marketing authorization holders are required to continue submitting data on authorized medicines in the XEVPRM format to fulfill their regulatory obligations under Article 57 of Regulation 726/2004.
For further details, refer to:
- Data submission on authorized medicines (Article 57)
Marketing authorization holders and national competent authorities are advised to:
- Synchronize their systems with the released terminologies for referentials, organization, and substance data.
- Request the registration of any new or missing controlled vocabulary necessary for medicinal product data submission.
- Commence structuring their product data according to the guidelines outlined in chapter 2 of the EU IG.
PMS implementation plan
EMA and the European medicines regulatory network have reached an agreement for a phased implementation strategy comprising three key stages:
- Product Data Preparatory Phase: This phase is centered on preparing the target operating model. It commenced with the introduction of the RMS and the OMS in 2017 and is set to extend through 2022 (refer to the PMS contribution to TOM in 2022 above).
- PMS Implementation – Step 1: This step involves certain aspects that EMA is still deliberating upon, such as whether to maintain the two-step approach (CAP & Fast Healthcare Interoperability Resources (FHIR) message first; non-CAPs second) and its implications on the process outlined in the EU Implementation Guide’s (EU IG) chapter 3.