
Nitrosamine “USFDA Guidelines: Managing Nitrosamine Contaminants in Human Medications”
Nitrosamine Earlier today (September 4, 2024), the Center for Drug Evaluation and Research of the U.S. Food and Drug Administration issued an updated version of the guidance document titled “Control of Nitrosamine Impurities in Human Drugs.” This second revision provides detailed recommendations for both active pharmaceutical ingredient (API) producers and pharmaceutical manufacturers on identifying and mitigating unacceptable levels of nitrosamine impurities in drug products.
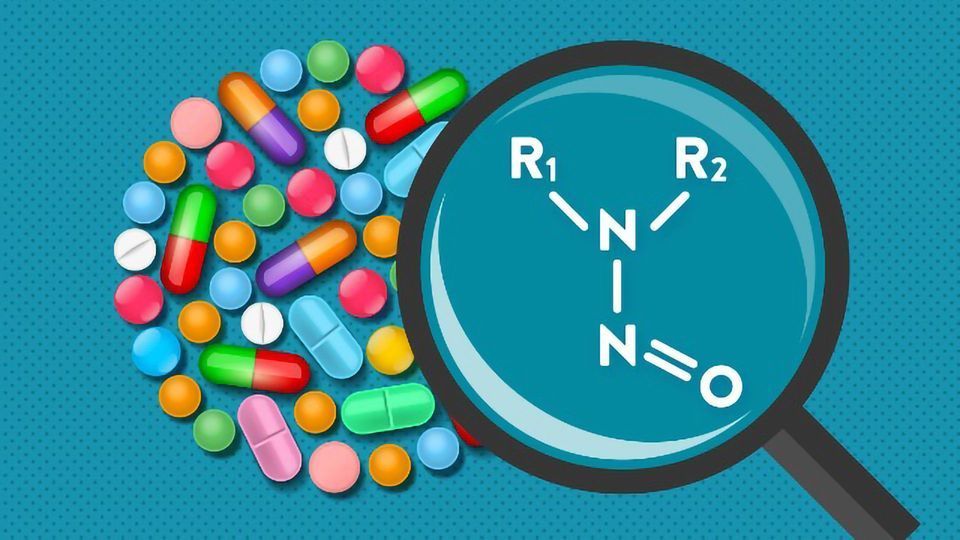
Nitrosamines refer to a group of compounds characterized by a nitroso group attached to an amine (R1N(-R2)-N=O). These compounds are known to be highly genotoxic in various animal models, and some are considered probable or possible human carcinogens according to the International Agency for Research on Cancer. Nitrites, which are common nitrosating agents, have been detected in many excipients at concentrations in the parts per million (ppm) range.
According to ICH M7(R2), an Acceptable Intake (AI) limit is defined as the level of a mutagenic impurity in drug substances and products that corresponds to an estimated increased cancer risk of one additional case in 100,000 individuals, assuming daily exposure over a lifetime (70 years).
The guidance identifies two primary categories of nitrosamine impurities: small-molecule impurities, which are structurally distinct from active pharmaceutical ingredients (APIs), and nitrosamine drug substance-related impurities (NDSRIs), which resemble APIs but are typically specific to each API. The document addresses potential causes of nitrosamine impurities, methods for their detection, and strategies to prevent or minimize their presence. These strategies include conducting risk assessments, performing testing, and implementing controls among other measures.
The guidance also includes recommendations for alternative bioequivalence methods for manufacturers and applicants who opt to reformulate their products to reduce nitrosamine impurities.
The recommendations in this guidance are applicable to:
- All chemically synthesized active pharmaceutical ingredients (APIs).
- Drug products containing chemically synthesized APIs or fragments, including biological products with synthesized fragments.
- Drug products that are at risk due to additional factors outlined in the guidance (see sections III.B., C., and D.).
- Semisynthetic and fermentation products that are structurally similar to chemically synthesized APIs and thus at risk.
Drug Products in Development and Under FDA Review Nitrosamine
Presubmission Stage
Prospective applicants should evaluate the risk of nitrosamine impurities early in the development process, ideally before conducting in vivo tests such as bioequivalence (BE) studies. The FDA advises that a scientifically sound risk assessment for nitrosamine impurities in active pharmaceutical ingredients (APIs) and proposed drug products should be completed, with confirmatory testing performed as needed, before the original application submission. If confirmatory testing or risk assessment results are not available at the time of the initial submission, they can be provided later through an amendment. It is important to submit such amendments promptly after the initial filing to avoid delays in the application review process.
Post-Submission Stage

For applications under review, it is essential that applicants quickly carry out risk assessments and inform the FDA if confirmatory testing detects nitrosamine levels exceeding the recommended acceptable intake (AI) limits. According to section 505(d)(3) of the FD&C Act, the FDA can refuse to approve an application if it finds that the manufacturing methods, facilities, or controls are inadequate to ensure the drug’s quality and purity. The FDA requires that a new drug application (NDA) includes a comprehensive chemistry, manufacturing, and controls (CMC) section, detailing manufacturing, packaging procedures, and in-process controls to maintain drug quality and purity. The FDA aims to resolve issues collaboratively with applicants during the review cycle and adheres to review goals set by the Prescription Drug User Fee Act and the Generic Drug User Fee Amendments.
Reporting Changes to Mitigate Nitrosamine Impurities
Applicants must report any changes made to prevent or reduce nitrosamine impurities according to FDA regulations (§§ 314.60, 314.70, 314.96, 314.97, and 601.12). Those with pending applications should update their submissions through an amendment following §§ 314.60 and 314.96.
- Reporting Changes by API Manufacturers If an API Drug Master File (DMF) holder alters the manufacturing process due to risk assessment and confirmatory testing results, they must submit an amendment to the DMF and notify all drug product manufacturers or applicants referencing the DMF (both pending and approved). Such changes should also be reported in the application per §§ 314.60, 314.70, 314.96, 314.97, and 601.12. For APIs manufactured by the applicant and not covered by a DMF, changes must be reported in the application as per the relevant regulations. Any reprocessing or reworking of batches containing nitrosamines should be documented in the DMF or application. If multiple synthetic routes are used, they should be clearly identified in the DMF with separate codes, and the removal of the original process should be scheduled and communicated. If a timely update is not feasible, a new DMF should be submitted for the revised process.
- Reporting Changes for Reformulated Drug Products To address nitrosamine impurities, reformulation strategies such as adding antioxidants or pH modifiers may be employed. If confirmatory testing reveals nitrosamines in a drug product, reformulation should be implemented to mitigate impurity levels. Changes must comply with applicable regulations, and formulation changes should be confirmed to meet specifications throughout the product’s shelf life. Manufacturing changes associated with reformulation must be reviewed and approved by the quality control unit. Significant changes, such as reformulation, typically require prior approval supplements as outlined in §§ 314.70(b)(1) and 314.97(a). Reformulation involving new excipients is considered a Level 3 change, often necessitating additional bioequivalence studies. Applicants with pending applications should submit amendments with relevant information. Data demonstrating continued compliance with specifications and bioavailability requirements must accompany reformulation changes. Applicants are encouraged to discuss formulation changes with the FDA prior to submission and should include stability and bioequivalence studies to support the change. Recommendations for stability testing follow ICH guidelines and FDA guidance documents on immediate-release solid oral dosage forms and changes in chemistry, manufacturing, and controls.
While nitrosamines are generally unlikely to form during the production of most APIs, it is important for all manufacturers of chemically synthesized APIs to implement measures to minimize the presence of these impurities where they might occur. API manufacturers should assess the risk of nitrosamine contamination in their manufacturing processes and perform thorough risk evaluations. If there is a potential for nitrosamine impurities, it is crucial to conduct sensitive and properly validated confirmatory testing of batches to ensure their safety.