
What is Bioavailability and Bioequivalence Study?
What is Bioavailability and Bioequivalence Study? Bioavailability refers to the proportion of a drug from a given dose that enters the bloodstream and the speed at which it reaches systemic circulation. Assessing bioavailability involves measuring drug concentrations in plasma or blood following standardized administration protocols, documented over time. These protocols are vital in early-stage drug development for clinical trials. The data obtained are then utilized in subsequent bioequivalence studies, which aim to differentiate between two pharmaceutical products containing the same active ingredient. If two formulations of a drug demonstrate therapeutic equivalence, they may be deemed interchangeable.
The field of pharmacokinetics examines how the concentration of a drug or its metabolites changes over time in the human or animal body after administering a pharmaceutical product. Bioequivalence studies are conducted to determine whether two different formulations of a drug exhibit comparable biological
effects when administered in vivo. When two pharmaceutical products are considered bioequivalent, it implies they are essentially identical in terms of their effects. Bioequivalence is typically established when the absorption rate and extent of a drug administered at the same molar dose are not significantly different from those of a reference product.
Various methods can be employed to evaluate equivalence, including: What is Bioavailability
- Comparative bioavailability studies, which measure the active drug substance in a readily accessible
biological fluid like plasma. - Comparative clinical trials.
- Comparative pharmacodynamic studies conducted in human subjects.
Bioavailability and bioequivalence studies are essential to ensure that a test drug, which is pharmaceutically equivalent to a generic or reference drug, maintains therapeutic equivalence. The Central Drugs Standard Control Organization (CDSCO) holds the primary responsibility of maintaining uniform standards of quality, efficacy, and safety across pharmaceutical products. Bioequivalence is crucial to establish the clinical equivalence and interchangeability of various products containing the same active ingredients but marketed by different licensees. Applicants seeking approval for new drugs under schedule Y must provide bioavailability and bioequivalence data, which primarily focus on drug release from the pharmaceutical dosage form and subsequent absorption into the systemic circulation.
Comparative bioavailability, also known as relative bioavailability, involves comparing two pharmaceutical dosage forms based on their relative rate and extent of absorption. In certain cases, such as comparing a rapidly absorbed elixir to a more slowly absorbed capsule, the bioavailability may vary significantly. However, in other instances, such as comparing a tablet to a capsule, the bioavailability may be quite similar or may differ only slightly. The formula for comparative bioavailability is expressed as:
Comparative bioavailability = (AUCpo × Doseiv) / (AUCiv × Dosepo).
Absolute bioavailability pertains to the active pharmaceutical ingredient’s ability to enter the systemic circulation, with the fraction of drug absorbed falling within the range of 0 to 1. A value of F at zero signifies no absorption of the drug, while a value of F equal to 1 indicates complete absorption into the systemic circulation. The quantity of drug that reaches the systemic circulation is directly correlated with the area under the curve (AUC), and the fraction of drug absorbed is determined by comparing the
respective AUCs of the test product to that of the same dose administered intravenously. Mathematically, absolute bioavailability is represented as the ratio of the AUC of the orally administered product to the AUC of the intravenously administered product. Thus, the formula for absolute bioavailability is expressed as: Absolute bioavailability = AUCpo / AUCiv.
1.1 In bioequivalence studies, various types of research methodologies are necessary to assess the equivalence between a test drug and a reference drug. These methodologies typically include:
For specific medications, determining in vivo equivalence is accomplished through either a bioequivalence investigation or a comparative clinical pharmacodynamic analysis. In the case of immediate-release oral formulations designed for systemic effects, there are several challenging
factors such as a narrow therapeutic window, a steep dose-response curve, nonlinear pharmacokinetics, presystemic elimination, and unfavorable physicochemical characteristics. These physicochemical issues encompass problems like drug solubility and instability, metastable transformation, poor permeability, particularly in drugs with similar chemical structures or formulations where excipients significantly outnumber active ingredients.
Medications administered through routes other than oral or parenteral formulations, those with sustained release formulations for systemic absorption, fixed-dose combination products with systemic effects,
pharmaceutical products not intended for systemic absorption, and those designed for non-systemic use require careful study. In such cases, the concept of bioequivalence may not be applicable, necessitating comparative clinical or pharmacodynamic investigations to establish equivalence.
Bioequivalence studies serve to establish correlations between early and late-stage clinical trial formulations, formulations used in clinical trials and stability assessments, as well as clinical trial
formulations and the final marketed drug products. Each comparison involves the new formulation or manufacturing method as the test drug, while the prior formulation serves as the reference drug.
1.2 When bioequivalence studies are unnecessary.
In certain scenarios, bioequivalence studies may not be deemed necessary if the similarity between a test drug and a reference drug is apparent without the need for extensive documentation. This is particularly the case when the test drug is in a gaseous form, or when it is intended for parenteral administration
(such as subcutaneous, intramuscular, or intravenous) as an aqueous solution, containing identical drug concentrations and excipients in comparable amounts. Similarly, bioequivalence studies may be waived for oral solutions with identical drug dosages and lacking excipients known to influence gastrointestinal absorption, as well as for ophthalmic or topical products prepared as aqueous solutions with identical active ingredients and excipient concentrations. Additionally, for powder formulations intended for
reconstitution into a solution, bioequivalence may not be required if the resulting solution meets the criteria outlined above. Similarly, for inhalation or nasal spray formulations, bioequivalence studies may be waived when the test is conducted using the same device as the reference drug.
2. The planning and execution of pharmacokinetic investigations involve careful consideration of study design and methodology.
2.1 Object Study
The design of a bioavailability study determines its protocol. A protocol aimed at estimating pharmacokinetic parameters differs from a bioequivalence study, which evaluates the test formulation against a standard formulation.
2.2 Study structure design
The primary aim of experimental design is to minimize variables and eliminate bias. When conducting in vivo bioavailability studies, several key points should be considered:
- Assessing the nature of the reference drug and the dosage form under examination.
- Evaluating the benefit-risk ratio associated with human testing.
- Ensuring availability of appropriate analytical methods.
- Clarifying the scientific questions that need to be addressed.
Bioavailability studies are impacted by numerous factors including age, gender, health status, dietary habits, physical and mental conditions, body weight of volunteers, experimental setup, timing of
administration and sampling, analytical techniques employed, and the compartment model used for estimating pharmacokinetic parameters. Hence, careful consideration of these factors is essential in study design.
The design of bioavailability studies should enable the differentiation of formulation effects from other variables. For comparing two formulations, a two-period, two-sequence crossover design is preferred, with a duration ideally equal to or exceeding five half-lives of the substances being measured. Alternatively, for substances with very long half-lives and highly variable disposition, a parallel design may be considered.
The following sections will delve into various factors, with a focus on bioequivalence studies, but the principles discussed are applicable to straightforward bioavailability studies as well.
2.2.1 Parallel design
In a parallel study design, two different formulations are given to separate groups of volunteers. To mitigate bias, the formulations are randomly assigned to the volunteers. However, a significant drawback of this approach is the failure to account for intersubject variation. Research consistently demonstrates that intersubject variation often surpasses variation between formulations. Hence, in bioavailability or bioequivalence trials, a crossover design is typically favored to minimize the impact of intersubject
variability. This design is particularly suitable for drugs and their metabolites with prolonged elimination half-lives. Parallel studies generally exhibit fewer carryover effects or dropouts compared to crossover studies.
2.2.2 Crossover design
As per the guidelines set forth by the US Food and Drug Administration (USFDA), most bioequivalence investigations involve comparing a test drug with a standard reference drug among a cohort of healthy individuals aged 18 to 55 years. Each participant receives both treatments alternately in a crossover
manner, typically following a two-period, two-treatment crossover design. The treatment phases are separated by a washout period, typically lasting around a week, which is determined primarily by the drug’s half-life; a longer half-life necessitates a longer washout period.
In these studies, whether the volunteers receive the test or reference formulation is randomized, ensuring an equal distribution of subjects for each treatment in each period. For instance, in a two-treatment scenario, two groups, labeled as Group 1 and Group 2, are formed. Group 1 undergoes treatment in the sequence A and B, while Group 2 follows the reverse order, receiving treatments in the sequence B and A. A similar randomization process is employed for a three-treatment crossover design.
Variability is observed among individuals in terms of drug clearance, both within and between subjects. Intrasubject variability, typically around 15%, is notably smaller than intersubject variability, approximately 30%. Hence, crossover designs are generally preferred for conducting bioequivalence studies due to
their ability to mitigate the impact of intersubject variability.
Group Number Subjects within Group Treatment Periods Two-way Crossover Group I Group II
- Participants 1-6 Treated with A, then B
- Participants 7-12 Treated with B, then A Three-way Crossover Group I Group II Group III
- Participants 1-6 Treated with A, then C, then B
- Participants 7-12 Treated with B, then A, then C
- Participants 13-18 Treated with C, then B, then A Four-way Crossover Group I Group II Group III Group IV
- Participants 1-6 Treated with A, then B, then C, then D
- Participants 7-12 Treated with B, then D, then A, then C
- Participants 13-18 Treated with C, then A, then D, then B
- Participants 19-24 Treated with D, then C, then B, then A
In crossover study designs, treatments are evaluated within the same individual, aiming to minimize intersubject variability. These designs rely on three key statistical principles: randomization, replication, and error control. Randomization ensures unbiased allocation of treatments to subjects, while replication
involves employing multiple experimental subjects to enhance the reliability of estimates and provide more precise measurements of treatment effects. The required number of replicates depends on the magnitude of differences to be detected and the inherent variability of the data. Common crossover designs used in bioavailability trials include the Latin square crossover design and the balanced incomplete block design.
Period Treatment Comparison:
- In period I, treatment A was administered, followed by treatment B in period II.
- Conversely, in period I, treatment B was administered, followed by treatment A in period II.
- Treatment A was administered in period I and treatment C in period II.
- Treatment C was administered in period I and treatment A in period II.
- Period I involved treatment A, followed by treatment D in period II.
- Period I involved treatment D, followed by treatment A in period II.
- Treatment B was administered in period I and treatment C in period II.
- Treatment C was administered in period I and treatment B in period II.
- Treatment B was administered in period I and treatment D in period II.
- Treatment D was administered in period I and treatment B in period II.
- Treatment C was administered in period I and treatment D in period II.
- Treatment D was administered in period I and treatment C in period II.
A widely adopted method for conducting comparative bioavailability studies is to employ a randomized, balanced crossover design, such as the Latin square or complete crossover design. The balanced incomplete block design (BIBD) addresses several challenges encountered with the Latin square design. In BIBD, each subject receives no more than two formulations, with each formulation administered the same number of times. Additionally, each pair of formulations is administered together to the same number of subjects. For instance, in a BIBD with four formulations (A, B, C, and D), each subject receives two
formulations, each administered six times, and each pair of formulations is evaluated in two subjects (AB, AC, AD, BC, BD, and CD).
2.3 Washout period (days between 2 periods)
In a Latin square cross-over design, each participant is administered each formulation, and even in a Balanced Incomplete Block Design (BIBD), each participant receives two formulations at different occasions. The duration between these treatments is referred to as the “washout period,” necessary
for the complete elimination of the drug dosage to prevent carryover effects. In most cases involving drugs in cross-over designs, a minimum of 10 half-lives is recommended between treatments. This duration ensures the elimination of approximately 99.9% of the administered dose, with carryover limited to less than 0.1% from the initial treatment.
The number of washout periods required depends on the drug’s half-life and dosage. It also varies according to the type of cross-over design employed and the number of formulations being assessed. For instance, in the case of digitoxin with a half-life of 6–9 days, conducting a study evaluating four formulations using a Latin square design may extend beyond a year. However, many drugs exhibit half-lives ranging from 1 to 10 hours, making a one-week washout period commonly sufficient in reported studies.
It’s important to consider the elimination of drug metabolites from the body before initiating subsequent treatments.
2.4 Test Drug product & reference-standard
Test products in pharmaceutical development encompass new drug formulations crafted by pharmaceutical technologists or fresh dosage forms of existing drugs. These products undergo scrutiny against a reference standard recognized by the Food and Drug Administration (FDA) to secure marketing approval. Their evaluation aims to pinpoint the optimal dosage form or formulation for a new or existing
drug among various options exhibiting comparable performance in vitro tests. Furthermore, test products undergo comparison with recognized standards to assess their biological performance.
Generic products must undergo comparison with established dosage forms to validate their in vivo efficacy. Typically, the FDA acknowledges any innovator’s drug product as a reference standard—the innovator being the entity initially granted FDA approval for marketing the product in the country. Sometimes, multiple manufacturers may hold approval for the same drugs, allowing any of the approved drug products to serve as a reference standard. However, the FDA often requests the use of a single product as a reference to facilitate more straightforward data comparison.
Bioavailability studies predominantly focus on orally administered dosage forms, although alternative routes such as buccal, transdermal, and intramuscular administrations should also undergo assessment for their biological performance. The therapeutic effectiveness of these dosage forms hinges on the
drug’s absorption rate and extent from them. Oral dosage forms exhibit considerable performance variation due to intersubject and intrasubject differences
2.5 Single(one) Vs multiple (more than one) dose study design
If the intention is solely to assess bioequivalence of dosage forms, single dose studies suffice. This is because the relative bioavailability of most tablets and capsules can typically be assessed based on
a single dose, which often correlates with performance across multiple doses. For dosage forms intended for single dose administration to achieve therapeutic effects, such as analgesics for headache relief, single dose studies are adequate. However, certain formulations aimed at achieving specific drug release profiles, such as time-release products, enteric-coated preparations, and certain intramuscular injections, may necessitate multiple dose studies. Additionally, drugs subject to first-pass metabolism also warrant multiple dose studies for comprehensive evaluation.
2.6 Administration of Test drug products/Reference and time point sampling
The administration of drug products or formulations to subjects should follow a randomized approach. Subsequently, blood samples are collected from the subjects at predetermined time intervals. Due to the time required for sample collection from each subject, the total duration between the first and last
subject’s sample may vary by 10 to 20 minutes, depending on factors such as the number of subjects and technicians involved. Deviations from the scheduled sampling sequence could lead to significant differences in the actual duration of drug presence in the body and the designated sampling times for each subject. This variance in sample collection time could result in notable differences in observed drug concentrations in the blood, especially if treatments are administered sequentially under these conditions.
Assessing the bioavailability of a particular dosage form via a blood level study necessitates obtaining estimates for parameters such as the area under the serum concentration-time curve, peak plasma concentration (Cmax), and time of peak plasma concentration (Tmax). Hence, the frequency and duration of sampling are critical aspects of the study design and will vary depending on the drug. An adequate number of sampling points is essential for accurate evaluation of the area under the blood level curve, typically extending up to three to five half-lives of the drug. If the drug’s half-life is unknown, sampling should continue until approximately 1/10 or 1/20 of the peak levels are reached.
Urinary excretion studies come into play when measuring a drug in blood, plasma, or serum is not feasible or when ethical constraints prohibit sample collection over time. This method offers non-invasive sampling, typically resulting in higher drug concentrations in urine compared to serum, and provides a direct measure of the drug excreted. However, urinary excretion studies may not effectively estimate the absorption rate of rapidly absorbed drugs, and interference from metabolites could affect the accuracy of estimating unchanged drug levels in urine samples.
Sampling should span a sufficient duration to ensure that the area extrapolated from the last measured concentration to infinite time remains below 20% of the total AUC (area under the curve). AUC calculations may not be suitable for drugs undergoing enterohepatic recycling, where accurately calculating the terminal elimination rate constant is challenging. In such cases, a minimum of three
sampling points during the absorption phase, three to four points around Tmax, and four points during the elimination phase are recommended. The intervals between successive sampling points in the terminal elimination phase should not exceed the half-life of the study drug.
2.7 Selection of population (number of subjects)
A sufficient number of participants must be included in the study to account for potential withdrawals or dropouts. In the event of a participant withdrawal or dropout during the initial stages, it is permissible to replace them, provided that the substitute participant adheres to the original protocol and undergoes
testing under similar conditions as the withdrawn participant.
Determining the appropriate number of participants for a study involves several considerations:
- The significance level should be set at 0.05.
- The error variance associated with the key variables under investigation, as estimated from pilot experiments or previous studies, should be taken into account.
- The anticipated deviation from the reference drug, while still maintaining bioequivalence, needs to be considered.
- The study should have sufficient statistical power, typically greater than 80%, to detect any significant differences in the primary variables being studied
2.7.1 Selection acceptance criteria for Population (subjects/Volunteers)
The research should involve healthy adult participants, ensuring consistency among the study drugs. Both male and female subjects may participate, but their selection should align with the drug’s usage and safety guidelines. To reduce variability among participants, the study’s design should be standardized to the greatest extent possible and deemed acceptable.
2.7.2 fed & Fasting condition
Typically, a single dose study necessitates an overnight fast of at least 10 hours, followed by an additional fasting period of 4 hours post-dosing. In the case of multiple dose studies, a 2-hour fasting window before and after administration is deemed acceptable. For modified release products or drugs, assessment of Cmax and Tmax may require fed state studies alongside the standard fasting state bioavailability studies. During fed state studies, subjects are required to consume a high fat breakfast totaling 950–1000 KCals prior to dosing. This breakfast should consist of at least 50% fat calories, 15–20% protein calories, and the remainder from carbohydrates. A uniform standard diet representative of
the Indian subcontinent population should be adhered to. The high fat breakfast should be ingested approximately 15 minutes before dosing under fed state conditions.
2.8 Study-Conditions/food & posture-restriction
Monitoring various factors such as the study environment, dietary habits, hydration levels, posture after dosing, physical activity, and sampling schedules is essential during studies. These factors are outlined in the protocol, and it’s imperative to adhere to them at the conclusion of the study to ensure minimal
variability in the products being tested. Subjects are required to abstain from smoking, consuming alcohol, xanthine-containing foods, coffee, tea, beverages, and fruit juices at least 48 hours before the study begins.
2.9 Steady state studies are conducted under specific conditions outlined as follows:
- The drug possesses a prolonged terminal elimination half-life.
- Blood concentrations cannot reach a sufficient level after a single dose for an extended period.
- In cases where administering drugs with toxic or adverse effects to patients is ethically prohibited, yet they remain essential for therapy (e.g., cytotoxics).
- Evaluation of fluctuation in plasma drug concentration at steady state is necessary for modified or
sustained-release products. - Instances where the drug is prone to accumulation within the body.
- Examination of drugs with nonlinear pharmacokinetics, characterized by dose or time dependency.
- Importance of assessing the ratio of plasma concentration for individual drugs in combination products.
- Relevance for drugs that induce their metabolism.
- Evaluation of innovative enteric-coated preparations.
2.10 Analysis of biological/blood plasma samples
Typically, biological samples gathered according to the prescribed sampling procedure should be promptly analyzed following the study. However, it’s common for these samples to be stored for several days before undergoing analysis. Throughout storage, various factors such as chemical degradation and adsorption onto container walls can affect the integrity of the drug. Thus, proper storage of plasma samples holds significant importance in bioavailability studies. The analytical method employed for assessing the active ingredient responsible for therapeutic efficacy should be both selective and sensitive. Drugs subject to the first-pass effect may display varying ratios of unchanged drug to metabolite depending on absorption rates. In the analysis of blood and urine, a primary challenge lies in achieving quantitative extraction and subsequent separation of the intact drug from its principal metabolites, or even separating a mixture of multiple drugs from their metabolites.

2.11 Assessment Methods for bioavailability (BA)
Pharmacokinetic techniques are employed to evaluate the bioavailability of pharmaceutical products, establishing a direct correlation between drug concentration in bodily fluids and therapeutic effectiveness. Consequently, these approaches are often termed as indirect methods. Since the quantification of therapeutically active substances in biological fluids such as plasma and urine is highly
precise, data obtained from these sources offer the most reliable insights into bioavailability.
2.11.1 Pharmacokinetic(PK) methods/Indirect methods sheet
Plasma data serves as the predominant and widely accepted means for evaluating the bioavailability of a drug product. This method operates on the foundational premise that bioequivalent drug products exhibit nearly identical plasma concentration-time curves. Key parameters such as Tmax and Cmax signify the rate of drug absorption, whereas AUC reflects the extent of absorption.
Conversely, the urinary excretion method relies on the principle that the rate of drug excretion via urine correlates directly with its blood concentration. Hence, bioavailability can be computed by comparing the total amount of unchanged drug excreted in urine after administering test and standard formulations. However, urinary metabolite excretion data aren’t utilized for bioavailability estimation due to potential variations in drug metabolism across different bodily sites, including the gut and liver.
For bioequivalence assessment, the relative bioavailability should typically fall within an acceptance range of 0.80–1.25, considering a 90% confidence interval. In situations with a narrow therapeutic range, a tighter acceptance range might be necessary. In exceptional cases involving highly variable drugs, a broader acceptance range could be justifiable with appropriate clinical reasoning. Notably, the Cmax ratio, a measure of relative bioavailability, may exhibit more variability compared to the AUC ratio, hence allowing for a wider acceptance range. Nonetheless, the chosen range in the protocol should be
supported by considerations of safety and efficacy. Additionally, Tmax serves as an indicator of drug release, action, or potential adverse effects.
2.11.2 Pharmacodynamic(PD)methods/Direct methods
Pharmacodynamic techniques come into play when it’s not feasible to assess bioavailability through pharmacokinetic methods due to either the unavailability of a precise analytical technique for drug
measurement or when existing methods lack the necessary precision and reproducibility. Two primary pharmacodynamic approaches are employed to gauge bioavailability: one centered on acute pharmacological effects and the other on clinical responses. For accurately estimating the bioavailability of a drug product through acute pharmacological effects, certain criteria must be fulfilled. These include the presence of easily measurable responses such as heart rate, ECG, blood pressure, pupil diameter, etc., and the availability of a well-established dose-response curve.
2.12 (ANOVA) Analysis of variance & Statistical analysis of the data
Owing to inherent biological and experimental discrepancies, discerning whether observed differences stem from chance or actual variations in administered treatments is imperative. Statistical methodologies are employed to assess pharmacokinetic data, aiming to delineate diverse sources of variation and
quantify their respective impacts, thereby isolating the primary observations of interest. One such widely utilized technique in bioavailability testing is Analysis of Variance (ANOVA), particularly in the context of crossover designs.
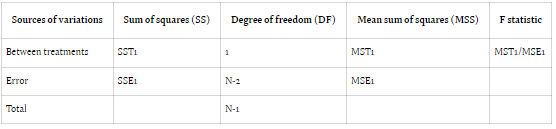
In ANOVA, variance is attributed to subjects, treatment periods, and the treatments themselves. The conventional null hypothesis (H0: μT = μR) tests for bioequivalence between pharmaceutical products, while the alternative hypothesis (H1: μT ≠ μR) indicates bioequivalence, with μT and μR denoting
the expected mean bioavailability of the test and reference drugs, respectively.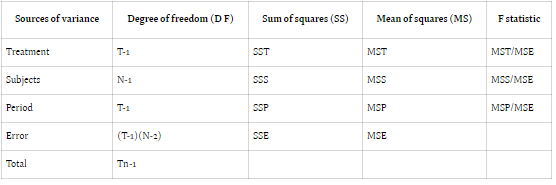
Bioavailability studies typically adopt one of two designs: Design 1 (parallel) or Design 2 (crossover). Design 1 allocates subjects into two treatment groups, each receiving a distinct treatment, while Design 2 involves a crossover arrangement, wherein each subject undergoes both treatments with a washout
period between them. In parallel designs, treatment-related variability is emphasized, whereas crossover designs account for treatment, subject, and period variability to minimize overall variance.
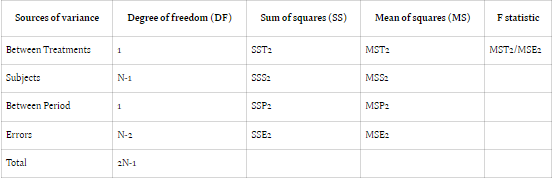
Comparing error sum of squares (SSE) in Design 1 (SSE1) with Design 2 (SSE2) reveals an equality between the two. However, the mean sum of square errors (MSE) in Design 1 (MSE1) typically surpasses that of Design 2 (MSE2), assuming equivalent degrees of freedom for SSE in both designs. This discrepancy suggests greater error variability in parallel group designs compared to crossover designs.
2.13 Attributes to be examined in bioequivalence investigations
Evaluation of bioavailability and bioequivalence studies hinges on monitoring the concentrations of active drug substances in plasma over time. There are scenarios where measuring active or inactive metabolites becomes necessary. These situations arise when drug concentrations are too low for accurate measurement in the biological matrix, when there are limitations with the analytical method, when dealing with unstable drugs, or drugs with a very short half-life. Racemates should be assessed using an achiral assay method. In bioequivalence studies, individual enantiomers must be measured if they demonstrate distinct primary efficacy, safety activity, pharmacodynamic, or pharmacokinetic characteristics, particularly with the minor enantiomer. Key pharmacokinetic parameters for both products and steady state, such as Cmax, Tmax, AUC0-t, AUC0-∞, AUC0-τ, Cmin, and degree of fluctuation, are derived from the plasma concentration-time profile.
2.14 Testing of Bioavailability(BA) & Bioequivalence (BE)
Bioavailability and bioequivalence testing involve comparing two formulations, such as a new formulation and an already marketed brand drug. These tests are typically carried out using experimental designs like parallel or crossover designs, often in healthy volunteers and occasionally in patients. During these studies, the formulation is administered under controlled conditions, and plasma samples are taken at regular intervals to measure the concentration of the drug or its metabolites in plasma or urine. However, in some cases, comparing drug concentrations in the blood may not be feasible. The collected plasma
concentration data are then analyzed to determine various pharmacokinetic parameters such as AUC, Cmax, Tmax, and absorption lag time (Tlag). It’s important to conduct bioavailability studies at different doses, especially if the drug exhibits nonlinear pharmacokinetics. Besides data from bioequivalence studies, additional evidence may be required to meet regulatory standards, including validation of analytical methods and in vitro-in vivo correlation studies.
2.15 Bioequivalence Acceptance Criteria.
Establishing bioequivalence for AUC, Tmax, and Cmax typically requires a 90% confidence interval falling within the range of 80–125%. A one-sided t-test with a 5% level of significance is used to test the null hypothesis of bioequivalence. In bioavailability investigations, narrower limits are preferred for drugs with a narrow therapeutic index, significant dose-related toxicity, steep dose-response curve, and nonlinear pharmacokinetics within the therapeutic dose range. However, a broader acceptance range might be permissible with robust clinical justification. In instances of supra bioavailability, reformulation of
the drug product is necessary, followed by another bioequivalence study. The application of the new formulation is essential to support clinical trial data, particularly for dosage recommendations. Nevertheless, such formulations are typically not considered therapeutically equivalent to the existing reference drug.